Le energie relative delle subshell elettroniche sono state calcolate per atomi in prossimità di $ Z = 20 $ ( J. Chem. Educ . , 1994 , 71 (6), 469) e il risultato è sorprendente:
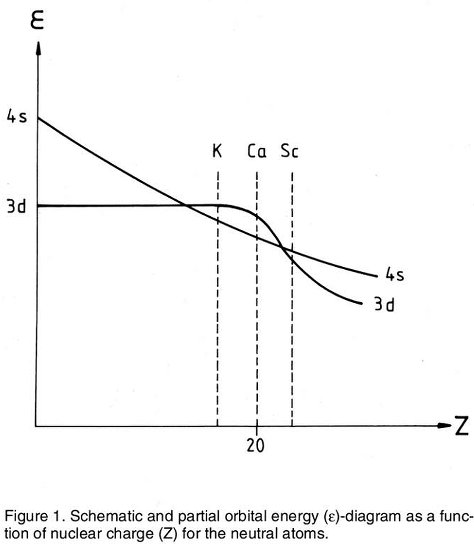
Guardando questo grafico, la configurazione elettronica dello scandio dovrebbe in effetti essere $ \ ce {1s ^ 2 2s ^ 2 2p ^ 6 3s ^ 2 3p ^ 6} $ $ \ color {blue} {\ ce {3d ^ 3}} $ per ridurre al minimo le energie orbitali! Il grafico non mostra l'energia della sottoshell $ \ ce {4p} $ , ma si trova leggermente al di sopra di entrambe le curve mostrate. Tuttavia, c'è un effetto importante che non viene considerato qui, che sono le repulsioni interelettroniche destabilizzanti. Gli elettroni in una stessa subshell tendono a respingersi di più (repulsione intrasubshell) rispetto agli elettroni in diverse subshell (repulsione intersubshell). Risulta (e nessuno può davvero spiegare qualitativamente perché, vedi sotto) la repulsione è tale che in un atomo con $ Z = 21 $ span >, la configurazione $ \ ce {[Ar] 4s ^ 2 3d ^ 1} $ ha un'energia inferiore rispetto ad altri potenziali candidati, come $ \ ce {[Ar] 3d ^ 3} $ o $ \ ce {[Ar] 4s ^ 1 3d ^ 2} $ . Teoricamente, le configurazioni dispari $ \ ce {[Ar] 4s ^ 2 4p ^ 1} $ o $ \ ce {[ Ar] 4s ^ 1 3d ^ 1 4p ^ 1} $ avrebbe energie di repulsione interelettroniche inferiori rispetto allo stato fondamentale osservato, ma in questi casi le repulsioni interelettroniche abbassate non compensano la necessità di popolare l'energia superiore $ \ ce {4p} $ subshell. Il confronto di diverse possibili configurazioni di elettroni e la minimizzazione congiunta dell'energia orbitale e delle energie di repulsione degli elettroni non sembra essere qualcosa che può essere fatto senza ricorrere a calcoli pesanti o convalida sperimentale diretta.
Oltre ad alcuni materiali che ho collegato nei commenti, questo articolo dal blog di Eric Scerri (un chimico che si concentra sugli aspetti della periodicità, inclusa la distribuzione elettronica) afferma, riguardante la configurazione elettronica dello scandio rispetto a calcio e allo ione $ \ ce {Sc ^ 3 +} $ (enfasi mia):
Ciò equivale a dire che tutti tre degli elettroni finali entrano in $ \ ce {3d} $ ma due di loro vengono respinti in un orbitale energeticamente meno favorevole, il $ \ ce {4s} $ , perché il risultato complessivo è più vantaggioso per l'atomo nel suo insieme. Ma questo non è qualcosa che può essere previsto. Perché sono 2 elettroni, invece di uno o addirittura nessuno? In casi come il cromo e il rame, solo un elettrone viene spinto nell'orbitale $ \ ce {4s} $ . In un caso analogo della seconda serie di transizione, l'atomo di palladio, la competizione si verifica tra $ \ ce {5s} $ e $ \ ce {4d} $ orbitali. In questo caso nessuno degli elettroni viene spinto verso l'alto nell'orbitale $ \ ce {5s} $ e la configurazione risultante ha un guscio esterno di $ \ ce {[Kr] 4d ^ 10} $ .
Niente di tutto ciò può essere previsto in termini semplici da una regola empirica e così sembra quasi che valga la pena mascherare questo fatto affermando che la configurazione complessiva può essere prevista, almeno per quanto riguarda i casi in cui due elettroni vengono spinti in alto nell'orbitale s rilevante. A coloro a cui piace presentare un'immagine piuttosto trionfale della scienza è troppo ammettere che non possiamo fare queste previsioni. L'uso dello sciatto Aufbau sembra evitare questo problema poiché fornisce la configurazione complessiva corretta e quasi nessuno sente l'odore di un topo.
Quindi, sfortunatamente, sembra che anche se la maggior parte degli effetti che si combinano per produrre le configurazioni elettroniche osservate sono noti, non esiste un modo qualitativo per prevedere dove le configurazioni non corrisponderanno al principio di Aufbau o ai livelli di gli orbitali. Ho letto l'affermazione che il principio di Aufbau è decisamente sbagliato praticamente per ogni atomo rispetto al posizionamento dei livelli di energia orbitale, ma incredibilmente accade di prevedere la configurazione del guscio di valenza per la maggior parte degli atomi.