Non c'è nulla di banale nei calcoli MCSCF perché è difficile prevedere a priori quanto tempo richiederà un calcolo. Esistono equazioni ben definite per calcolare quanti determinanti
$$ D (n, N, S) = \ binom {n} {N / 2 + S} \ binom {n} {N / 2-S} $$
o funzioni di stato della configurazione (CSF)
$$ D (n, N, S) = \ frac {2S + 1 } {n + 1} \ binom {n + 1} {N / 2-S} \ binom {n + 1} {N / 2 + S + 1} $$
sarà richiesto per il tuo problema; $ n $ è il numero di orbitali attivi, $ N $ è il numero di elettroni attivi e $ S $ lo spin totale. Il costo di calcolo scalerà con questo numero e può essere ulteriormente ridotto con l'uso della simmetria del gruppo puntuale. (In realtà, a seconda del sistema e della proprietà di interesse, i calcoli senza simmetria possono essere privi di significato, come per gli stati eccitati e le loro energie.)
Queste equazioni dovrebbero chiarire che il costo cresce molto rapidamente, considerando che per CASSCF, viene eseguito un CI completo all'interno dello spazio attivo scelto. Questo non è ciò che rende i calcoli MCSCF complicati nella pratica. Ecco un grafico dell'energia totale dopo le prime 30 iterazioni.
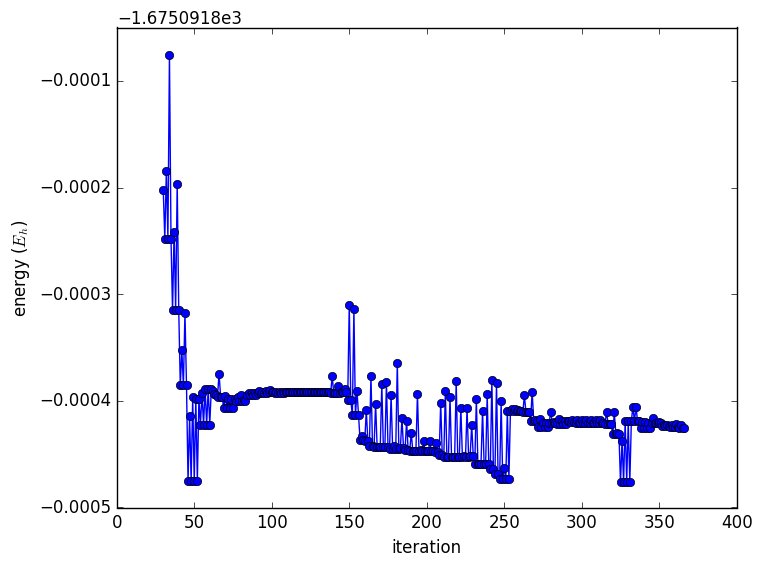
Questo perché i numeri di occupazione sono molto vicini a 0 / 1/2 e non cambiano con ogni iterazione. Si noti che i numeri di occupazione si sono invertiti e ripetuti ad ogni passaggio è Rot = 23,19 . L'orbitale 19 è all'interno dello spazio attivo, ma 23 è esterno. Questa è un'indicazione che gli orbitali nello spazio attivo non sono quelli corretti. Dal documento:
i 12 o 13 elettroni di valenza del neutro o dell'anione, rispettivamente, sono stati distribuiti tra 14 orbitali attivi di valenza: 3p di zolfo più 3d, 3d 'e 4 s orbitali di ferro. Con questo spazio attivo abbiamo scoperto che tutti gli orbitali correlati importanti sono inclusi nello spazio attivo.
MCSCF non è una scatola nera; è uno dei pochi metodi chimici quantistici rimasti che richiede l'intuizione chimica anche solo per iniziare. Questo è uno (12,14) spazio attivo; quello nei tuoi calcoli è (12,8). La procedura che utilizzo per eseguire i calcoli MCSCF è generalmente:
-
Converge un calcolo a riferimento singolo. Questo potrebbe essere HF, DFT, MP2, ecc. La solita raccomandazione è di formare orbitali naturali MP2, quindi controllare i numeri delle occupazioni. Se l'occupazione devia di più di 0,02 da un valore intero, probabilmente dovrebbe essere nello spazio attivo. Questo è in aggiunta alla considerazione degli elettroni e degli orbitali che volevi correlare all'inizio, insieme ai partner antilegame. Ad esempio, nell'etilene questa è probabilmente la coppia pi legame / antilegame, che porta a un (2,2), e per i complessi metallici 3d questo è qualunque sia il coordinamento (anti) legami presenti, con potenzialmente il doppio guscio d. Ho avuto un po 'di fortuna nell'usare gli orbitali DFT per complessi metallici, dove la teoria delle perturbazioni potrebbe essere una cattiva idea a causa di quasi degenerazioni o costi. Gli obiettivi qui sono confermare il tuo spazio attivo e fornire i migliori orbitali iniziali possibili per MCSCF.
-
Guarda i tuoi orbitali. Ovviamente, leggi il Analisi della popolazione Mulliken o Lowdin, ma per esperienza personale, ho colto molti potenziali errori in questo modo. Aiuta anche quando si visualizza come gli orbitali a singolo stato e riferimento singolo si trasformano negli orbitali naturali (potenzialmente medi di stato) che risultano dai calcoli MCSCF.
-
Riordina gli orbitali se necessario prima di eseguire MCSCF. Fatta eccezione per i casi più banali, gli orbitali che appartengono allo spazio attivo non sono gli orbitali che escono direttamente dal tuo calcolo a riferimento singolo. Se lo fossero, potresti non eseguire MCSCF per cominciare. Tendo anche a guardare ancora una volta gli orbitali riordinati prima di eseguire MCSCF per confermare di non aver commesso errori.
-
Leggi la serie precedente di orbitali (riordinati) in un calcolo MCSCF separato e incrocia le dita. Il monitoraggio dei numeri di occupazione del tuo spazio attivo è una buona guida per vedere come il calcolo sta convergendo.
-
Fai qualsiasi post-elaborazione di cui hai bisogno per il tuo progetto. Per me, si tratta di esaminare i numeri di occupazione finale, gli orbitali stessi e quindi eseguire MR-CISD o altri calcoli correlati a più riferimenti.
Eseguire una procedura di autooccupazione senza partire da orbitali precedentemente convergenti significa che 1. non hai alcuna garanzia sui contenuti del tuo spazio attivo, 2. parti da orbitali che sono vicini per qualità a Hartree-Fock e 3. il calcolo deve fare molto più lavoro nell'ottimizzazione di entrambi gli insiemi di parametri variazionali (i coefficienti MO $ \ {C \} $ e i coefficienti CI $ \ {c \} $) poiché gli orbitali interni ed esterni (dove i coefficienti CI sono congelati) sono ancora lontani convergenza utilizzando un metodo di riferimento singolo.
Per esaminare parte di questo flusso di lavoro, ho eseguito un calcolo PBE0 / aug-cc-pVTZ (converge molto più velocemente dell'SCF prima di MP2) per osservare il MO di Lowdin popolazioni:
! pbe0 aug-cc-pvtz cc-pvtz / jk ri rijk tightscf usesym zora% output print [p_orbpopmo_l] 1 end * xyz 0 5Fe 0.000000 0.000000 0.000000S 1.960000 0.000000 0.000000 *
Gli indici MO sono S 3s: 14, Fe 4s: 20, Fe 3d: 15,16,17,18,19 e Fe 3d ': 33,34,36-40. L'occupazione automatica avrebbe dato ragione agli occupati, ma i virtuali sbagliati. Non sono sicuro di quale dei 7 MO virtuali che potrebbero essere il Fe 3d 'siano quelli corretti; probabilmente ci sono alcuni tentativi ed errori qui, ma questi dovrebbero essere visualizzati . Un altro punto è che ho esaminato solo l'ordine per gli orbitali con spin alfa; per casi difficili come i metalli di transizione, l'ordine spaziale per gli orbitali beta-spin può essere molto diverso. Il riordino degli orbitali alfa e beta spin potrebbe essere diverso. Ancora una volta, questo parla per esperienza.
La mostra finale sono gli orbitali naturali RI-MP2 / aug-cc-pVTZ che potrebbero entrare in un calcolo MCSCF in base ai loro numeri di occupazione:
N [15] (A2) = 1,97761944N [16] (A1) = 1,97753198N [17] (B2) = 1,97009578N [18] (B1) = 1,97009578N [19] (A1) = 1,03828526N [ 20] (A1) = 0,99215102N [21] (B1) = 0,98317529N [22] (B2) = 0,98317529N [23] (A2) = 0,02013554N [24] (B2) = 0,01947713N [25] (B1) = 0.01947713N [26] (A1) = 0.01816184
Per essere accurati, questo dovrebbe essere combinato con l'osservazione delle popolazioni Lowdin MO di questi, che potrebbe richiedere la lettura in un altro (SCF) calcolo e non eseguire alcuna iterazione.
Tuttavia, è ben noto che la convergenza di equazioni MCSCF è generalmente difficile. Nella mia limitata esperienza, DALTON, GAMESS e Molcas hanno meno problemi di ORCA. Non ho usato Molpro per MCSCF. Il programma scelto determina quali calcoli post-MCSCF possono essere eseguiti. L'algoritmo DIIS in ORCA, in particolare, può bloccarsi anche quando non dovrebbe. Se te lo puoi permettere, l'algoritmo di Newton-Raphson ( switchstep nr ), in cui l'orbitale hessiana dello spazio attivo viene calcolato ad ogni passaggio, funziona molto bene per forzare la convergenza una volta che SuperCI è stato fatto. ORCA rende facile iniziare con CASSCF e ha molte manopole, ma ha un motore integrato molto lento, non ti impedisce di fare cose stupide come GAMESS, e le funzionalità più avanzate sono scarsamente documentato. Anche il flusso di lavoro descritto sopra (vale a dire il tracciamento degli orbitali e la loro rotazione) diventa molto noioso, ma questo ha più a che fare con MCSCF in generale.
Ecco alcune buone risorse che descrivono aspetti sia pratici che teorici di esecuzione di calcoli MCSCF: 1, 2, 3, 4.