TL; DR La struttura cristallina di $ \ ce {[NEt4] 2 [InCl5]} $ [1] inizialmente pubblicata secondo le ulteriori indagini [3], non è valida. $ \ Ce {InCl5 ^ 2 -} $ ion non ha $ C_ \ mathrm {4v} $ simmetria, e la teoria VSEPR spiega praticamente la formazione di numerosi bipiramidali trigonali leggermente distorti $ \ ce {InCl5 ^ 2 -} $ - contenenti complessi secondo i più recenti esperimenti sulla struttura monocristallina.
Dall'altro lato, le lunghezze dei legami e l'indice di geometria si riferiscono al carattere dominante di ambiente piramidale quadrato , che in questo caso può essere dettato dall'imballaggio cristallino, verso il quale $ \ ce {[InCl5] ^ 2 -} $ è più sensato di $ \ ce {[SnCl5] -} $ a causa della notevole differenza di dimensioni ($ \ ce {[InCl5] ^ 2 -} $ è più voluminoso). In ogni caso, sembra che sia necessario un altro esperimento cristallografico (preferibilmente a temperature più basse per diminuire le dimensioni degli ellissoidi termici) per determinare gli angoli in modo più preciso.
Come consiglio generale, presta sempre attenzione ai valori $ R_1 $ e $ R_2 $ della struttura cristallina. Una regola pratica: un buon affinamento della struttura di piccole molecole dovrebbe portare a $ R_1 < 0,05 $ e $ R_2 < 0,12 $.
Struttura cristallina di $ \ ce {[NEt4] 2 [InCl5 ]} $, determinato nel 1969 [1], è stato successivamente criticato in diverse pubblicazioni, coinvolgendo principalmente l'analisi aggiuntiva della vibrazione di simmetria degli spettri Raman per $ \ ce {MX5 ^ 2 -} $ anioni [2]. È stato stabilito che $ \ ce {InCl5 ^ 2 -} $ ion non ha $ C_ \ mathrm {4v} $ simmetria (ma mantiene $ C_ \ mathrm {2v} $) e che il cristallo potrebbe non essere centrosimmetrico.
I risultati sono riassunti in [3]:
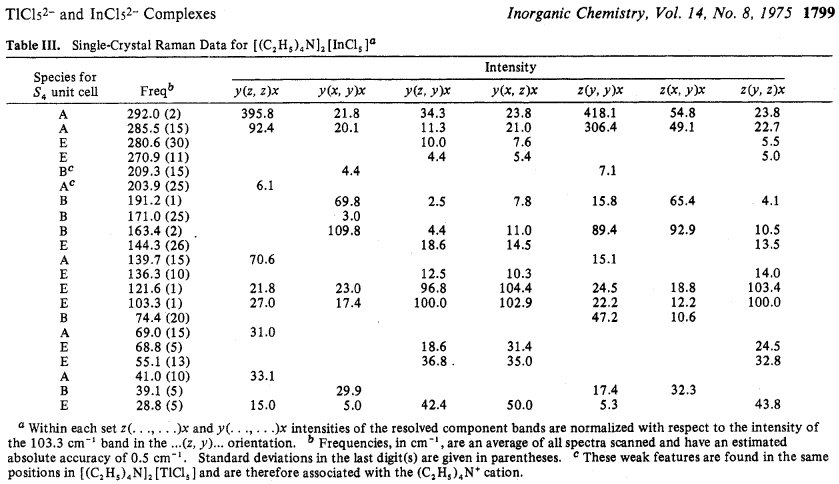
Deconvoluzione dei dati monocristallini è stato nuovamente eseguito con l'aiuto
dei dati sulla bassa temperatura, rivelando $ 11 $ $ \ ce {InCl5 ^ 2 -} $ bande comprese tra $ 300 $ e $ \ pu {100 cm-1} $. Due allungamenti A sovrapposti sono chiaramente evidenti a $ 292 $ e $ \ pu {286 cm-1} $, ma nessun allungamento B è evidente. I due allungamenti di simmetria E previsti per l'anione di un sito $ C_2 $, ma non per un sito $ C_4 $, sono evidenti a $ 281 $ e $ \ pu {271 cm-1} $. [...] Con l'aiuto di dati a bassa temperatura, le quattro modalità E previste per $ \ ce {InCl5 ^ 2 -} $ nei siti $ C_2 $ si trovano a $ 144 $, $ 136 $, $ 122 $ e $ \ pu {103 cm-1} $ nei dati monocristallini. Con questa reinterpretazione dello spettro non è più necessario ipotizzare arbitrariamente la presenza di un modo reticolare in questa regione.
In sintesi, i dati vibrazionali per i sali di tetraetilammonio di $ \ ce {InCl5 ^ 2- } $ e $ \ ce {TlCl5 ^ 2 -} $ indicano che questi complessi risiedono su siti privi della piena simmetria $ C_4 $. La conclusione più semplice dai dati vibrazionali è che gli ioni $ \ ce {InCl5 ^ 2 -} $ hanno una simmetria $ C_2 $ locale, che sarebbe incoerente con la determinazione della struttura precedente [1]. [...]
La struttura di $ \ ce {[(C2H5) 4N] 2 [InCl5]} $, come riportato in precedenza, consisteva in $ \ ce {InCl5 ^ 2 -} $ correlati centrosimmetricamente ioni situati intorno alle posizioni c di Wyckoff di $ C_4 $ simmetria nel gruppo spaziale $ C_ \ mathrm {4h} -P4 / n $. Coassiali con gli ioni $ \ ce {InCl5 ^ 2 -} $ sono disordinati $ \ ce {(C2H5) 4N +} $ anche situati attorno alle posizioni c Wyckoff. Gli N atomi dei due rimanenti ioni $ \ ce {(C2H5) 4N +} $ nella cella unitaria occupano i siti Wyckoff b correlati centrosimmetricamente della simmetria $ S_4 $, il catione nel suo insieme è nuovamente disordinato. Ci sono due caratteristiche di disturbo di questa soluzione e del suo successivo affinamento: il disordine degli ioni $ \ ce {(C2H5) 4N +} $ e la significativa densità elettronica residua nella regione degli atomi basali di Cl del anione .
È inoltre dimostrato che per il modello ordinato l'interpretazione cristallografica più soddisfacente coesiste con la simmetria $ C_2 $ invece che con $ C_4 $. Una correlazione delle simmetrie dei punti consentite implica il passaggio da $ P4 / n $ a $ P \ bar {4} $ spazio gruppo, riducendo così il fattore $ R $ ($ R_1 $) da $ 9.5 \% $ a $ 6.7 \% $.
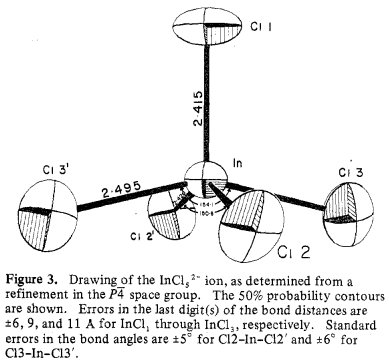
Durante il raffinamento in $ P \ bar {4} $, piccoli ma significativi cambiamenti si verificano nel piano basale di $ \ ce {InCl5 ^ 2 -} $ ion . Gli atomi basali di Cl si sono spostati di $ \ pu {-0,5 A} $ dalla loro quadruplice posizione simmetrica in $ P4 / n $, portando a variazioni nelle distanze di legame e negli angoli attorno all'atomo In (Figura 3) che sono qualitativamente sufficienti per spiegare i risultati vibrazionali.
Indice di geometria $ \ tau_5 $ per $ \ ce {[InCl5]} $ fragment può essere utilizzato per determinare l'ambiente di coordinamento formale come segue:
$$ \ tau_5 = \ frac {\ beta - \ alpha} {60 ^ \ circ}, $$
dove $ \ alpha $, $ \ beta $ - i due maggiori angoli di valenza del centro di coordinamento ($ \ angle \ ce {Cl - In - Cl} $, $ \ alpha < \ beta $).
\ begin {align} \ begin {cases} \ tau_5 & = 0 \ qquad & \ text {square pyramidal geometry} \\\ tau_5 & = 1 \ qquad & \ text {trigonal bipyramidal geometry} \ end { case} \ end {align}
Per la struttura $ \ ce {[NEt4] 2 [InCl5]} $ originale rifinita in [3] ho preso i valori angolari dai loro materiali supplementari. Si è scoperto che cambiare il gruppo spaziale in $ P \ bar {4} $ risulta ancora nel carattere dominante della geometria piramidale quadrata di $ \ ce {[InCl5] ^ 2 -} $, anche se bisogna ricordare che l'asse di rotazione dell'ordine più elevato è $ C_2 $, non $ C_4 $:
$$ \ alpha = (150.6 \ pm 5.0) ^ \ circ, \ beta = (154.1 \ pm 6.0) ^ \ circ, \ bar {\ tau_5} = \ frac {154,1 ^ \ circ - 150,6 ^ \ circ} {60 ^ \ circ} = 0,06; \ tau_5 \ in [0,04; 0.24] $$
L'articolo [4] menziona che per le strutture isoelettroniche $ \ ce {[SnCl5] -} $ - continenti con vari cationi sono state riportate solo geometrie trigonali-bipiramidali e suggerisce una maggiore influenza dell'impaccamento dei cristalli su $ \ ce {[InCl5] ^ 2 -} $ geometria. Ciò può anche essere spiegato dalla maggiore flessibilità del frammento $ \ ce {MX5} $ in $ \ ce {[PPh4] 2 [InCl5]} $ rispetto a $ \ ce {[PPh4] [SnCl5]} $ [5] dovuto a una lunghezza media del legame maggiore: $ d (\ ce {In - Cl}) = \ pu {2.5 A} $, $ d (\ ce {Sn - Cl}) = \ pu {2.3 A} $.
Per quanto riguarda i dati sperimentali ausiliari, tutte le strutture cristalline compreso il frammento $ \ ce {[InCl5] ^ 2 -} $ determinato negli ultimi due decenni supportano completamente la teoria VSEPR che predice la simmetria $ D_ \ mathrm {3h} $ bipiramidale trigonale ( con un'eccezione intermedia di $ \ tau_5 \ approx 50 \% $).
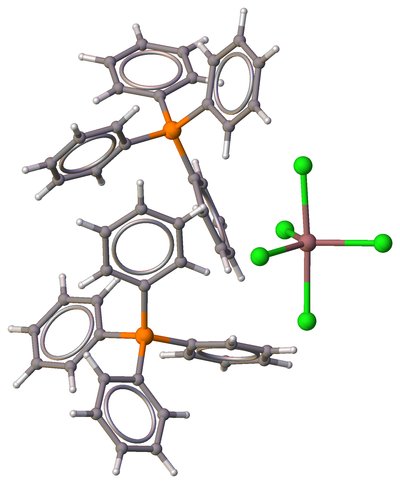
bis (Tetraphenylphosphonium) pentacloroindato (III) [4], geometria bipiramidale trigonale: $$ \ alpha = 124,24 ^ \ circ , \ beta = 174,74 ^ \ circ, \ tau_5 = \ frac {174,74 ^ \ circ - 124,24 ^ \ circ} {60 ^ \ circ} = 0,84 $$
$ \ color {#EEEEEE} { \ Large \ bullet} ~ \ ce {H} $; $ \ color {# 909090} {\ Large \ bullet} ~ \ ce {C} $; $ \ color {# FF8000} {\ Large \ bullet} ~ \ ce {P} $; $ \ color {# 1FF01F} {\ Large \ bullet} ~ \ ce {Cl} $; $ \ color {# A67573} {\ Large \ bullet} ~ \ ce {In} $.
-
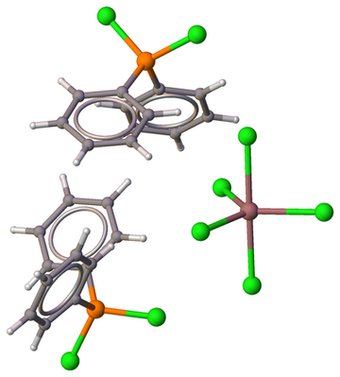
bis (difenildiclorofosfonio) pentacloroindato (III) [6], geometria bipiramidale trigonale: $$ \ alpha = 120,93 ^ \ circ, \ beta = 179,29 ^ \ circ, \ tau_5 = \ frac {179,29 ^ \ circ - 120,93 ^ \ circ} {60 ^ \ circ} = 0.97 $$
$ \ color {#EEEEEE} {\ Large \ bullet} ~ \ ce {H} $; $ \ color {# 909090} {\ Large \ bullet} ~ \ ce {C} $; $ \ color {# FF8000} {\ Large \ bullet} ~ \ ce {P} $; $ \ color {# 1FF01F} {\ Large \ bullet} ~ \ ce {Cl } $; $ \ color {# A67573} {\ Large \ bullet} ~ \ ce {In} $.
-
tetrakis ($ \ mu_3 $ -Selenido) -tetrakis ($ \ mu_2 $ -1,5-bis (difenilfosfino) pentano) -deca-oro (I) pentacloroindato (III) [7], geometria intermedia:
$$ \ alpha = 138,45 ^ \ circ, \ beta = 166,50 ^ \ circ, \ tau_5 = \ frac {166,50 ^ \ circ - 138,45 ^ \ circ} {60 ^ \ circ} = 0,47 $$
$ \ color {#EEEEEE} {\ Large \ bullet} ~ \ ce {H} $; $ \ color {# 909090} {\ Large \ bullet} ~ \ ce {C} $; $ \ color {# FF8000} {\ Large \ bullet} ~ \ ce {P} $; $ \ color {# 1FF01F} {\ Large \ bullet} ~ \ ce {Cl} $; $ \ color {# FFA100} {\ Large \ bullet} ~ \ ce {Se} $; $ \ color {# A67573} {\ Large \ bullet} ~ \ ce {In} $; $ \ color {# FFD123} {\ Large \ bullet} ~ \ ce {Au} $.
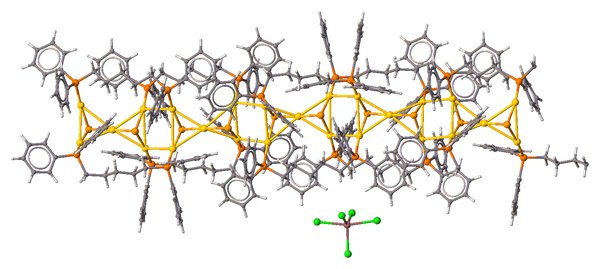
-
tetrakis ($ \ mu_2 $ -chloro) -cloro-tetrakis (trifenilfosfina-P) -di-rame (I) -indium (III) tetraidrofurano solvato [8], geometria bipiramidale trigonale (sebbene gli autori si riferiscano ad essa come a "quasi "coordinazione piramidale quadrata basata su un singolo $ \ ce {In - Cl} $ distanza di legame $ (d (\ ce {In-Cl_ \ mathrm {ap}}) = \ pu {2.36 A}) $, più breve $ d (\ ce {In-Cl_ \ mathrm {eq}}) = \ pu {2.42 A}) $: $$ \ alpha = 123.90 ^ \ circ, \ beta = 172.89 ^ \ circ, \ tau_5 = \ frac { 172,89 ^ \ circ - 123,90 ^ \ circ} {60 ^ \ circ} = 0,82 $$
$ \ color {#EEEEEE} {\ Large \ bull et} ~ \ ce {H} $; $ \ color {# 909090} {\ Large \ bullet} ~ \ ce {C} $; $ \ color {# FF8000} {\ Large \ bullet} ~ \ ce {P} $; $ \ color {# 1FF01F} {\ Large \ bullet} ~ \ ce {Cl} $; $ \ color {# C88033} {\ Large \ bullet} ~ \ ce {Cu} $; $ \ color {# A67573 } {\ Large \ bullet} ~ \ ce {In} $.
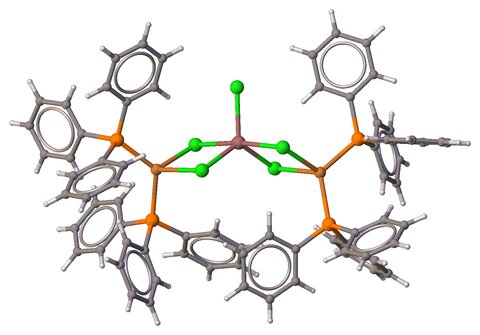
Bibliografia
- Brown, DS; Einstein, F. W. B .; Tuck, D. G. Inorganic Chemistry 1969 , 8 (1), 14-18. DOI 10.1021 / ic50071a004.
- Adams, D. M .; Smardzewski, RR Journal of the Chemical Society A: Inorganic, Physical, Theoretical 1971 , 714. DOI 10.1039 / j19710000714.
- Joy, G .; Gaughan, A. P .; Wharf, I .; Shriver, D. F .; Dougherty, J. P. Inorganic Chemistry 1975 , 14 (8), 1795-1801. DOI 10.1021 / ic50150a011.
- Bubenheim, W .; Frenzen, G .; Müller, U. Acta Crystallographica sezione C 1995 , 51 (6), 1120–1124. DOI 10.1107 / S0108270194011789.
- Müller, U .; Siekmann, J. F. Acta Cryst C 1996 , 52 (2), 330–333. DOI 10.1107 / S0108270195011073.
- Taraba, J .; Zak, Z. Inorganic Chemistry 2003 , 42 (11), 3591–3594. DOI 10.1021 / ic034091n.
- Olkowska-Oetzel, J .; Sevillano, P .; Eichhöfer, A .; Fenske, D. European Journal of Inorganic Chemistry , 2004 (5), 1100-1106. DOI 10.1002 / ejic.200300774.
- Zhang, X.-Z .; Song, Y.-W .; HuiWu, F .; Zhang, Q.-F. Zeitschrift für Naturforschung B 2007 , 62 (6). DOI 10.1515 / znb-2007-0605.