Approccio:
L'approccio adottato in questa proposta di sintesi del cloramfenicolo si basa in gran parte sulla sintesi di Greene del paclitaxel, utilizzando la metodologia di diidrossilazione proposta da Sharpless.
Sebbene non particolarmente elegante (I sperava in una sorta di idrogenazione asimmetrica per impostare gli stereocentri), arriva al prodotto in 7 passaggi rispettabili (inclusa l'impostazione di entrambi gli stereocentri, come richiesto), ed evita di dover introdurre protezione / direzione gruppi in qualsiasi fase.
Sintesi diretta:
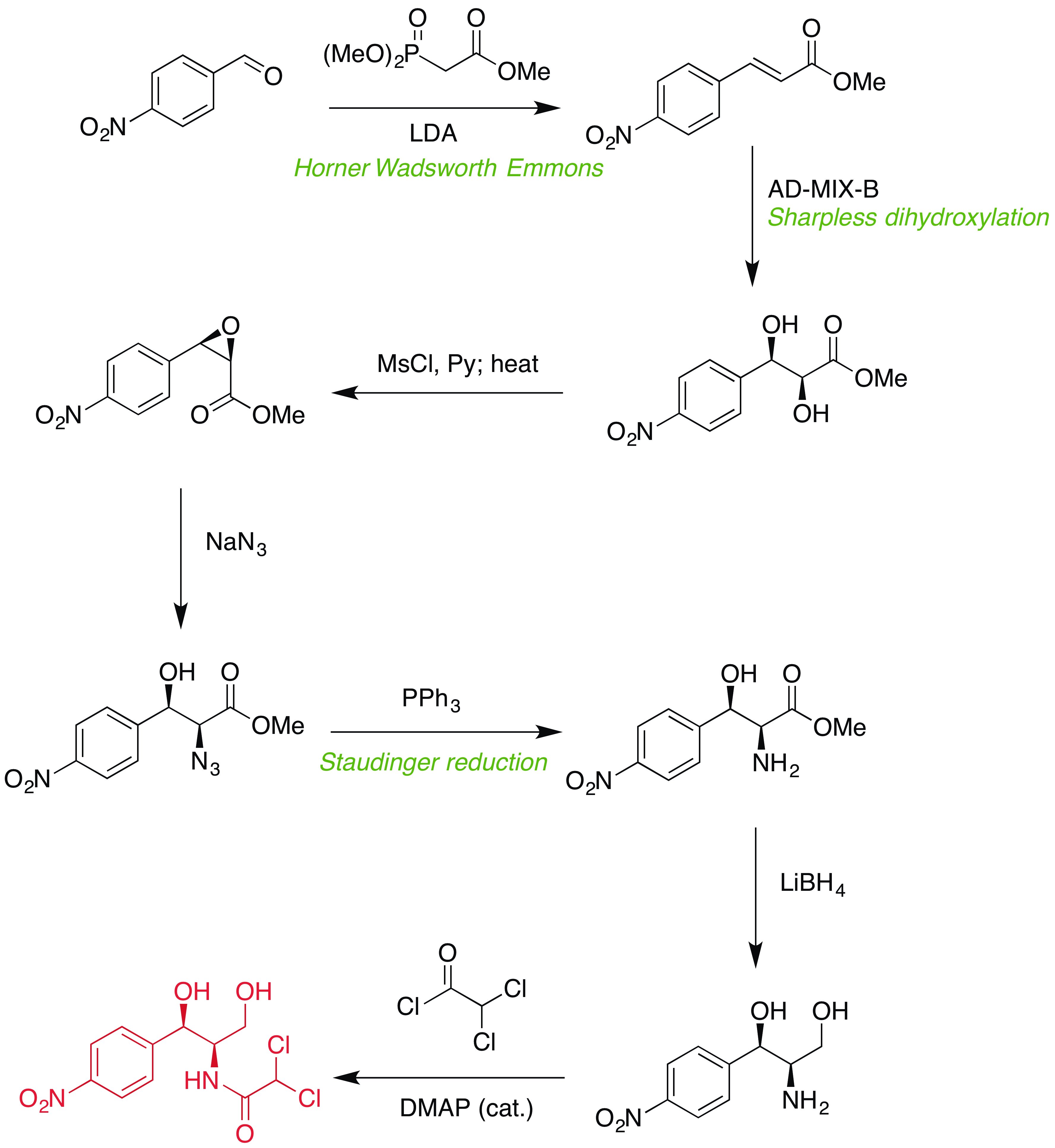
Passaggio 1: olefinazione di Horner Wadsworth Emmons
Il primo passo in questa sintesi proposta è un'olefinazione di Horner Wadsworth Emmons per introdurre l '(E) -alchene richiesto per la diidrossilazione pianificata. Senza il gruppo nitro, la reazione esatta è stata eseguita molte volte e con una buona selettività, quindi non c'è motivo di presumere che questo HWE sarebbe particolarmente impegnativo (il gruppo nitro rende chiaramente povero l'elettrone aldeidico).
Passaggio 2/3: diidrossilazione Sharpless, sequenza di epossidazione
Con la formazione di (E) -alchene, può avvenire una diidrossilazione asimmetrica Sharpless utilizzando AD-MIX-B (prendendo l'arile gruppo come "grande" e l'estere come "medio"). Questo passaggio (senza nitro) è presente nella sintesi del paclitaxel di Greene (sopra) e inizialmente dà l'82% di ee, sebbene una singola ricristallizzazione del prodotto porti questo a >95%.
Una volta ricristallizzato, il trattamento con il cloruro di mesile e il successivo riscaldamento (stessa pentola) forniscono l'epossido necessario.
Passaggio 4/5: apertura e riduzione dell'epossido
Ora arriva il passaggio un po 'dubbio ....
Nella sintesi del paclitaxel, l'epossido viene aperto con sodio azide nella posizione benzilica (previsto, l'anello aromatico può stabilizzare la carica positiva parziale in via di sviluppo qui), tuttavia nel nostro caso, abbiamo un incredibilmente elettroni che ritirano il gruppo nitro nella posizione 4, interrompendo completamente qualsiasi stabilizzazione nella posizione benzilica.
Sulla base di questa mancanza di stabilizzazione benzilica, potremmo ragionevolmente presumere che lo farà si svolgono con la regioselettività opposta al caso del paclitaxel. Ci sono alcune prove per questo in letteratura, (incluso dove c'è un estere presente sull'altro lato, ma purtroppo non si usa l'azide come nucleofilo), e un rapido sfogliare un libro di testo organico dirà Tu che SN2 adiacente ai carbonili è generalmente veloce.
Supponendo che funzioni bene, una riduzione di Staudinger converte l'azide nell'ammina secondaria con ritenzione di stereochimica.
Passaggio 6: Riduzione di esteri
Il boroidruro di litio è un reagente delicato e selettivo, in grado di ridurre gli esteri presenti in varie altre funzionalità, compresi i gruppi nitro. Uno svantaggio di questo è che il boroidruro di litio è significativamente più lento rispetto alle sue controparti (per non parlare di più costoso), ma l'uso di LAH o di altre specie di idruro altamente reattive provocherebbe reazioni collaterali che coinvolgono il gruppo nitro.
Passaggio 7: formazione del legame ammidico
Il cloruro acilico richiesto è disponibile in commercio e dovrebbe fare la reazione richiesta al posto delle formazioni estere possibilmente competitive (LP azoto più elevato in energia) .
Conclusione:
7 passaggi complessivi a partire dalla 4-nitrobenzaldeide disponibile in commercio. La maggior parte dei passaggi ha una buona precedenza su sistemi simili (ignorando l'apertura dell'epossido, che può / non può essere fatale), e tutte le reazioni proposte possono (e sono state) dimostrate possibili su larga scala (di nuovo, ignorando l'epossido apertura, ma la TMS-azide viene spesso sostituita nel caso di sintesi di processi che coinvolgono l'azide).