Vorrei eseguire il backup della risposta di Klaus con alcuni risultati della Teoria quantistica degli atomi nelle molecole (QTAIM), basati su un calcolo DF-BP86 / def2-SVP. Si noti che questi sono risultati, ottenuti senza considerare la solvatazione o le fasi condensate. Credo che dimostrino ancora un punto valido nel caso della teoria della struttura elettronica.
Ho rivisitato questa domanda per rispondere a un'altra domanda simile. Pur mettendo più impegno in questo, mi sono reso conto che le strutture qui trattate sono in realtà stati di transizione. Ciò non significa che i problemi risolti siano invalidati. Anche se esistono solo per brevissimi istanti, esistono ancora e devono essere considerati.
In o -metilanilina puoi vedere chiaramente il $ \ ce intramolecolare suggerito {H} $ bond. La distanza $ \ mathbf {d} (\ ce {NH}) = 239.3 ~ \ mathrm {pm} $ è solo di poco inferiore alla somma dei raggi di van der Waals, $ \ mathbf {r} (\ ce {N} ) = 155 ~ \ mathrm {pm} $, $ \ mathbf {r} (\ ce {H}) = 110 ~ \ mathrm {pm} $, ma anche trascurarlo è sbagliato. Anche se questa interazione esiste solo per periodi di tempo molto brevi, significa comunque che stabilizza questo stato. Tuttavia non sarà l'elemento dominante.
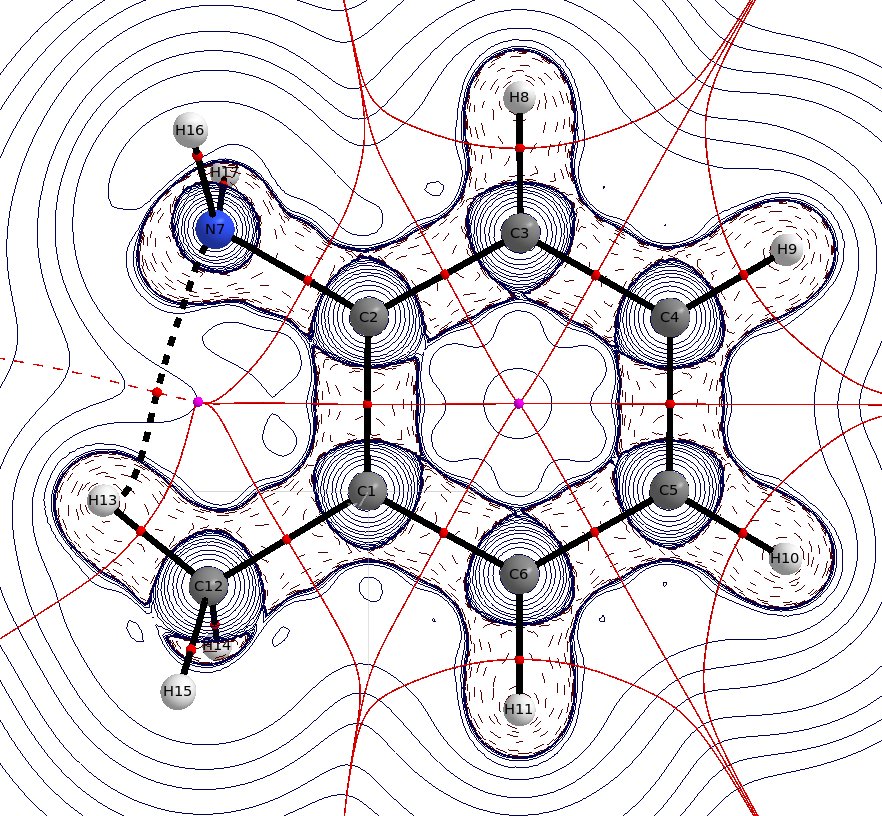
(Distribuzione laplaciana, le linee blu continue indicano l'esaurimento della carica $ \ nabla ^ 2 \ rho<0 $, le linee blu tratteggiate indicano l'accumulo di carica $ \ nabla ^ 2 \ rho>0 $, Le sfere rosse sono punti critici del legame, le sfere viola sono punti critici dell'anello, le linee nere sono percorsi di legame, le linee rosse sono superfici a flusso zero)
Gli effetti sterici sono solitamente effetti elettronici o dispersivi sotto mentite spoglie, quindi anche loro si riferiscono a un legame idrogeno intramolecolare. L'obbligazione $ \ ce {NH} $ media è solo di circa $ \ mathbf {d} _ \ text {av.} (\ Ce {NH}) \ approx99-105 ~ \ mathrm {pm} $.
La solvatazione il punto fatto da user4604 dovrebbe ancora essere considerato.
Questa interazione deve abbassare l'affinità protonica e / o l'affinità acida di Lewis e quindi diminuisce anche la basicità. Non tanto perché i suoi effetti elettronici stabilizzano una particolare conformazione, ma soprattutto, rendendo la coppia solitaria non disponibile per brevi periodi di tempo.
Puoi anche analizzare questo per o -acido metilbenzoico e qui l'effetto cambia direzione.
Nell'acido benzoico sono già presenti dei legami idrogeno intramolecolari degli orto idrogeni, uno di questi è ancora presente nel caso sostituito. La distanza $ \ mathbf {d} (\ ce {O-H_ {o '}}) = 226.2 ~ \ mathrm {pm} $ è solo di poco inferiore alla somma dei raggi di van der Waals, $ \ mathbf {r} (\ ce {O}) = 151 ~ \ mathrm {pm} $, $ \ mathbf {r} (\ ce {H}) = 110 ~ \ mathrm {pm} $, ma trascurarlo sarebbe comunque sbagliato. La distanza $ \ mathbf {d} (\ ce {O-H_ {Me}}) = 210.8 ~ \ mathrm {pm} $ è significativamente più breve della somma dei raggi di van der Waals. Puoi di nuovo vedere l'interazione tramite un percorso di legame e suonare i punti critici. Nella geometria ottimizzata, la parte metilica è leggermente ruotata, dando luogo a due interazioni equidistanti. La rotazione di questo gruppo può essere considerata come una rotazione libera a temperatura ambiente.
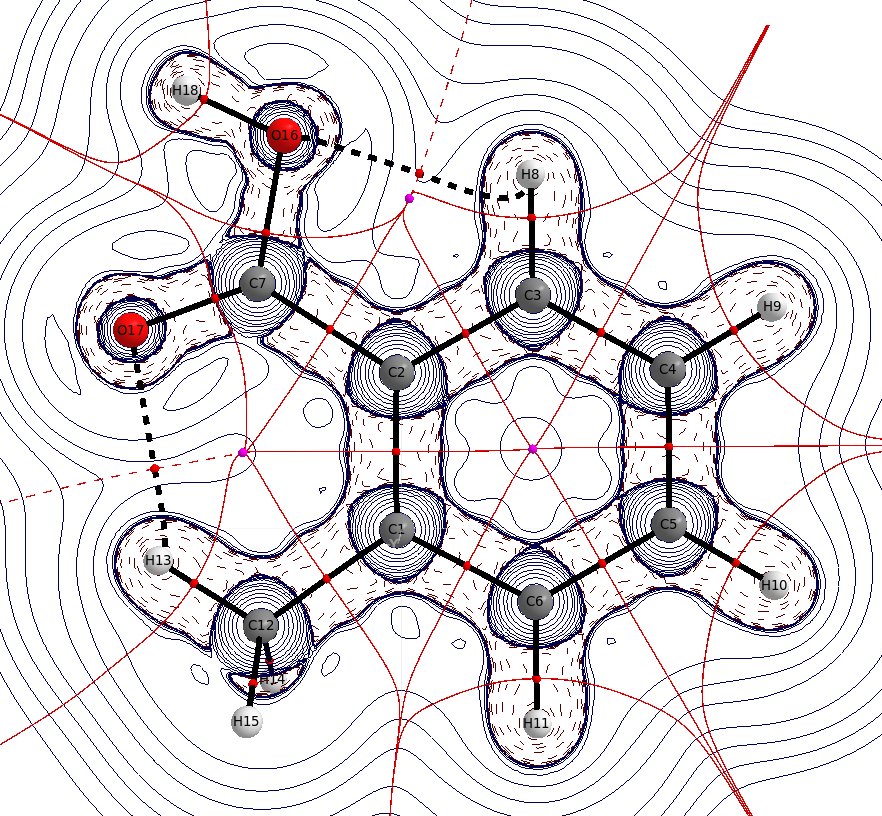
È interessante notare che il punto critico di legame del $ \ ce {O-H_ {acid}} $ è quasi nella posizione del protone, il che indica anche che la maggior parte della densità elettronica appartiene già all'ossigeno.
È anche ovvio che la concentrazione di carica in questo legame è significativamente inferiore rispetto a qualsiasi altro $ \ ce {EX} $ bond, che può indicare un'obbligazione debole.