La risposta di Klaus Warzecha risponde praticamente alla tua domanda. Ma so che questo argomento è più facile da capire se supportato da alcune immagini. Ecco perché prenderò la stessa strada di Klaus per spiegare il concetto alla base del perché l'assorbimento nei sistemi coniugati è spostato a lunghezze d'onda più elevate, ma fornirò alcune immagini lungo la strada.
In una catena o anello di carbonio coniugato sistema puoi pensare agli $ \ ce {C} $ atoms come $ \ text {sp} ^ {2} $ - ibridato. Quindi, ogni carbonio ha 3 $ \ text {sp} ^ {2} $ orbitali che usa per formare $ \ sigma $ legami e 1 $ \ text {p} $ orbitale che viene usato per formare $ \ pi $ legami. sono gli $ \ text {p} $ orbitali responsabili della coniugazione e le loro combinazioni secondo il modello LCAO sono la parte interessante poiché l'HOMO e il LUMO del sistema saranno tra gli orbitali molecolari formati dal coniugato $ \ text { p} $ orbitali.
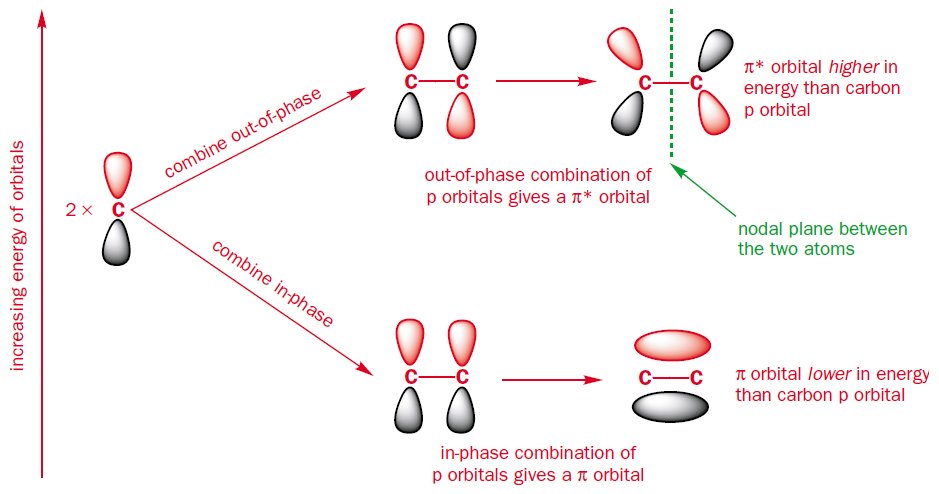
Per cominciare prendi l'etene, il sistema $ \ pi $ più semplice, essendo composto da soli 2 atomi di carbonio. Quando combini due orbitali atomici ottieni due orbitali molecolari. combinando gli orbitali $ \ text {p} $ in fase o fuori fase. La combinazione in fase ha un'energia inferiore rispetto agli orbitali $ \ text {p} $ originali e la combinazione fuori fase è maggiore in energia rispetto agli orbitali $ \ text {p} $ originali. La combinazione in fase rappresenta l'orbitale molecolare di legame ($ \ pi $), mentre la combinazione fuori fase rappresenta l'orbitale molecolare antilegame ($ \ pi ^ {*} $).
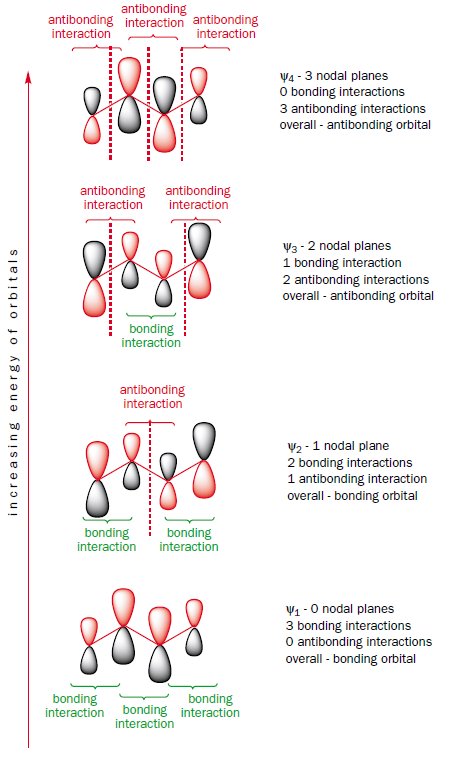
Ora, cosa succede quando allunghi il sistema coniugato combinando due frammenti di etene? Si arriva al butadiene. Il butadiene ha due $ \ legami pi $ e quindi quattro elettroni nel sistema $ \ pi $. In quali orbitali molecolari si trovano questi elettroni? Dato che ogni orbitale molecolare può contenere due elettroni, solo i due orbitali molecolari più bassi di energia sono riempiti. .
In $ \ Psi_1 $, l'orbitale di legame a più bassa energia, gli elettroni sono distribuiti su tutti e quattro gli atomi di carbonio (sopra e sotto il piano) in un orbitale continuo. C'è legame tra tutti gli atomi. Gli altri due elettroni sono in $ \ Psi_2 $ .Questo orbitale ha interazioni di legame tra gli atomi di carbonio 1 e 2, e anche tra 3 e 4 ma un'interazione antilegame tra i carboni 2 e 3. Nel complesso, in entrambi gli orbitali $ \ pi $ occupati ci sono elettroni tra i carboni 1 e 2 e tra 3 e 4, ma l'interazione di antilegame tra i carboni 2 e 3 in $ \ Psi_2 $ annulla parzialmente l'interazione di legame in $ \ Psi_1 $. Questo spiega perché tutti i legami nel butadiene non sono gli stessi e perché il legame medio è più simile a un legame singolo mentre i legami finali sono doppi legami. Se osserviamo attentamente i coefficienti su ciascun atomo negli orbitali $ \ Psi_1 $ e $ \ Psi_2 $, si può vedere che l'interazione di legame tra gli atomi di carbonio centrali in $ \ Psi_1 $ è maggiore di quello di antilegame in $ \ Psi_2 $. Quindi il butadiene ha un carattere di doppio legame tra i carboni 2 e 3, il che spiega perché esiste la leggera barriera alla rotazione attorno a questo legame.
Puoi costruire gli orbitali molecolari di butadiene combinando gli orbitali molecolari dei due frammenti di etene in fase e fuori fase.
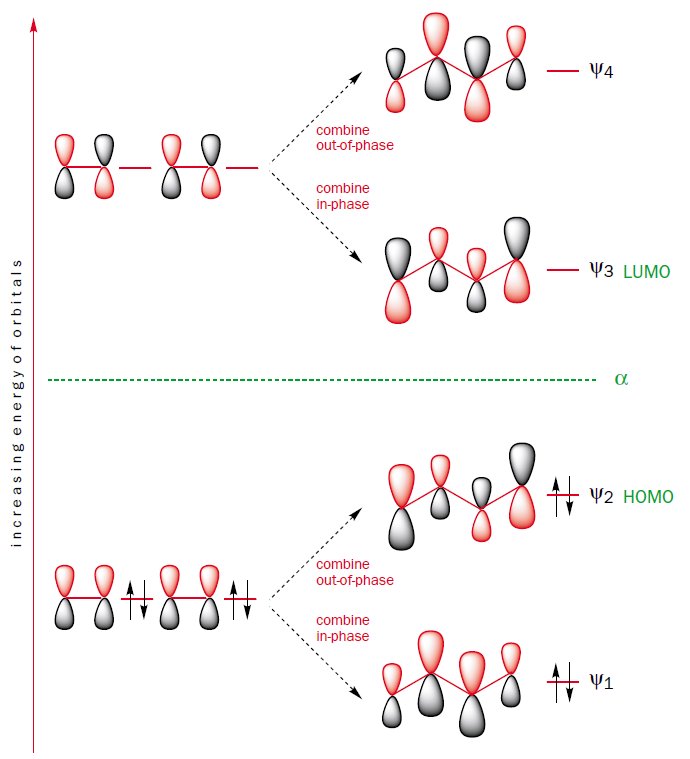
Questo metodo di costruzione mostra anche perché l'HOMO-LUMO gap di butadiene è più piccolo di quello di etene. L'orbitale molecolare $ \ Psi_2 $, che è l'HOMO del butadiene, è la combinazione sfasata di due orbitali $ \ pi $ di etene, che sono gli HOMO dell'etene. , l'HOMO del butadiene ha un'energia più alta dell'HOMO dell'etene. Inoltre, l'orbitale molecolare $ \ Psi_3 $, che è il LUMO del butadiene, è la combinazione in fase di due orbitali $ \ pi ^ {*} $ dell'etene , che sono i LUMO dell'etene, quindi il LUMO del butadiene ha un'energia inferiore al LUMO dell'etene.
Ne consegue che il gap energetico HOMO-LUMO è più piccolo nel butadiene rispetto all'etene e quindi il butadiene assorbe la luce con lunghezze d'onda maggiori dell'etene.
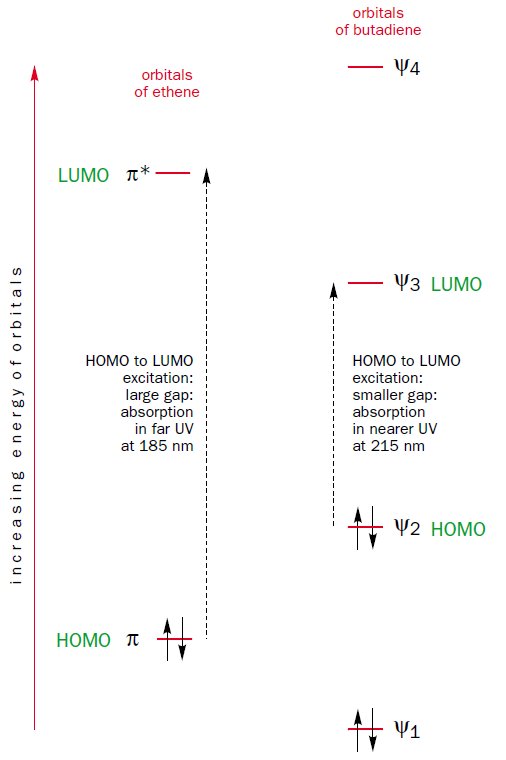
Se continui ad allungare il $ \ pi $ system aggiungendo più frammenti di etene vedrai che HOMO e LUMO si avvicinano sempre di più insieme più a lungo diventa il $ \ pi $ system.