Sfortunatamente GAMESS non è così facile da usare come altri pacchetti di chimica quantistica, quindi quando si tratta di eseguire calcoli non standard è necessario un po 'più di impegno.
Prima di tutto consiglio di ottimizzare i reagenti con stessi, in modo da avere un'idea in qualche modo valida di quale sarà la durata del legame al tuo livello di teoria. Dopo di che dovresti assemblare la tua migliore ipotesi per la struttura. In questo esempio ho scelto una separazione tra le molecole di circa 220 pm.
Le coordinate di questa struttura sono:
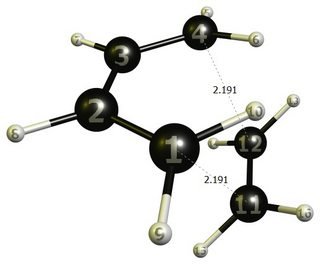
! DIENEC -,486294808 -0,431915730 -1.395523790C -1,166676574 0,550,50804 milioni -0.705577944C -1,166676574 ,550,50804 milioni 0.705577944C -,486294808 -,431915730 1.395523790H -,352988862 -,347708199 2.465648783H -,084717264 -1,289523730 0.864198908H -1,712490163 1,338,624758 millions 1.223514576H -1,712490163 1,338,624758 millions -1.223514576H -,352988862 -0.347708199 -2.465648783H -0.084717264 -1.289523730 -0.864198908! DIENOPHILEC 1.559305232 -0.085874351 -0.690651129C 1.559305232 -0.085874351 0.690651129H 1.514911897 -1.022640981 1.232100569H 1.603704743 0.851022589 1.232175888H 1.603708843 0.85102.932 si avvicinano a -1.532.532 H 1. . Qui vogliamo solo fissare due lunghezze di legame, 1 (C) -11 (C) e 4 (C) -12 (C). Se dovessimo fissare le coordinate di questi atomi, dovremmo anche fissare 1 (C) -4 (C) e 11 (C) -12 (C). Pertanto, un approccio migliore è utilizzare coordinate interne, in GAMESS viene utilizzato il gruppo di parole chiave $ ZMAT .
Dato che ce ne sono molti, è difficile impostarli. Utilizzeremo quindi la procedura automatizzata implementata. A partire da una prova a secco, ad esempio EXETYP = CHECK , in modo da poter verificare cosa fa effettivamente il programma. Dobbiamo definire $ 3 \ times N-6 = 42 $ coordinate interne, quindi NZVAR = 42 . Notare inoltre che in questo caso non usiamo la simmetria. A causa della natura della struttura dobbiamo aggiungere alcuni legami già manualmente, NONVDW (1) = 1,11, 4,12 . Questo è l'input risultante: $ CONTRL SCFTYP = RHF RUNTYP = ENERGIA EXETYP = CONTROLLO COORD = UNIQUE ICHARG = 0 MULT = 1 NZVAR = 42 $ END $ BASIS GBASIS = STO NGAUSS = 3 $ END $ ZMAT AUTO = .TRUE. DLC = .TRUE. NONVDW (1) = 1,11, 4,12 $ END $ GUESS GUESS = HUCKEL $ END $ DATA C6H10 C1 CARBON 6.0 -0.486294808 -0.431915730 -1.395523790 CARBON 6.0 -1.166676574 0.550508040 -0.705577944 CARBON 6.0 -0.486294808 -0.431915730 -1.395523790 CARBON 6.0 -1.166676574 0.550508040 -0.705577944 CARBON 6.0 -1.15080944 0.70944 CARBON 6.050 -0,431915730 1,395,52379 milioni IDROGENO 1,0 -0,352988862 -0,347708199 2,465648783 IDROGENO 1,0 -0,084717264 -1,289523730 0,864198908 IDROGENO 1,0 -1,712490163 1,338624758 1,223514576 IDROGENO 1,0 -1,712490163 1,338624758 -1,223514576 IDROGENO 1,0 -0,352988862 -0,347708199 -2,465648783 IDROGENO 1,0 -0,084717264 -1,289523730 -0,864198908 CARBONIO 6.0 1,559,305232 millions - 0.085874351 -0.690651129 CARBONIO 6.0 1.559305232 -0.085874351 0.690651129 IDROGENO 1.0 1 .514911897 -1.022640981 1.232100569 IDROGENO 1.0 1.603704743 0.851022589 1.232175888 IDROGENO 1.0 1.603704743 0.851022589 -1.232175888 IDROGENO 1.0 1.514911897 -1.022640981 -1.232100569> $ END
Il risultante file di output ausiliario .dat ha ora il gruppo che useremo da qui in avanti. E il file .log ci darà il valore esatto del coordinate che vogliamo congelare. -------------------- COORDINATE INTERNE -------------------- - - ATOMS - - COORDINATE COORDINATE NO. TIPO IJKLMN (BOHR, RAD) (ANG, DEG) -------------------------------------- -------------------------- 1 STRETCH 1 11 4.1406435 2.1911343 ... 10 STRETCH 4 12 4.1406435 2.1911343 ...
Ora possiamo modificare il nostro gruppo $ ZMAT di conseguenza. Sfortunatamente l'input diventa molto lungo, se vuoi mantenere una certa leggibilità. Nota che creiamo coordinate interne ridondanti, quindi dobbiamo aumentare anche il numero di NZVAR per leggerle tutte.
Tecnicamente non dobbiamo impostare la generazione delle coordinate su false , ma aiuta a ricordare cosa hai fatto se torni tra un mese circa, quindi AUTO = .FALSE. . Ora aggiungiamo le coordinate che vorremmo congelare con IFZMAT (1) = <list> e i valori corrispondenti FVALUE(1)=<list>.
$ CONTRL SCFTYP = RHF RUNTYP = OTTIMIZZA COORD = UNICO ICHARG = 0 MULT = 1 NZVAR = 89 $ END $ BASIS GBASIS = STO NGAUSS = 3 $ END $ ZMAT AUTO = FALSE. DLC = .TRUE. IZMAT (1) = 1, 1, 11, 1, 1, 10, 1, 1, 9, 1, 1, 2, 1, 2, 8, 1, 2, 3, 1, 3, 7, 1, 3 , 4, 1, 4, 6, 1, 4, 12, 1, 4, 5, 1, 11, 16, 1, 11, 15, 1, 11, 12, 1, 12, 14, 1, 12, 13 , 2, 1, 11, 15, 2, 1, 11, 16, 2, 1, 2, 3, 2, 1, 2, 8, 2, 1, 11, 12, 2, 2, 3, 7, 2 , 2, 1, 10, 2, 2, 1, 11, 2, 2, 1, 9, 2, 2, 3, 4,
2, 3, 2, 8, 2, 3, 4, 5, 2, 3, 4, 12, 2, 3, 4, 6, 2, 4, 3, 7, 2, 4, 12, 13, 2, 4, 12, 14, 2, 4, 12, 11, 2, 5, 4, 12, 2, 5, 4, 6, 2, 6, 4, 12, 2, 9, 1, 10, 2, 9, 1, 11, 2, 10, 1, 11, 2, 11, 12, 13, 2, 11, 12, 14, 2, 12, 11, 15, 2, 12, 11, 16, 2, 13, 12, 14, 2, 15, 11, 16, 3, 1, 2, 3, 4, 3, 1, 2, 3, 7, 3, 1, 11, 12, 14, 3, 1, 11, 12, 4, 3, 1, 11, 12, 13, 3, 2, 1, 11, 15, 3, 2, 1, 11, 16, 3, 2, 3, 4, 5, 3, 2, 3, 4, 12, 3, 2, 3, 4, 6, 3, 2, 1, 11, 12, 3, 3, 2, 1, 11, 3, 3, 4, 12, 13, 3, 3, 4, 12, 14, 3, 3, 4, 12, 11, 3, 3, 2, 1, 9, 3, 3, 2, 1, 10, 3, 4, 12, 11, 15, 3, 4, 12, 11, 16, 3, 4, 3, 2, 8, 3, 5, 4, 12, 11, 3, 5, 4, 12, 14, 3, 5, 4, 12, 13, 3, 5, 4, 3, 7, 3, 6, 4, 12, 11, 3, 6, 4, 12, 14, 3, 6, 4, 12, 13, 3, 6, 4, 3, 7, 3, 7, 3, 2, 8, 3, 7, 3, 4, 12, 3, 8, 2, 1, 9, 3, 8, 2, 1, 10, 3, 8, 2, 1, 11, 3, 9, 1, 11, 16, 3, 9, 1, 11, 15, 3, 9, 1, 11, 12, 3, 10, 1, 11, 12, 3, 10, 1, 11, 15, 3, 10, 1, 11, 16, 3, 13, 12, 11, 15, 3, 13, 12, 11, 16, 3, 14, 12, 11, 15, 3, 14, 12, 11, 16, IFZMAT(1)= 1, 1, 11, 1, 4, 12, FVALUE(1)= 2.1911343, 2.1911343, $END $GUESS GUESS=HUCKEL $END $DATA C6H10 C1 CARBON 6.0 -0.486294808 -0.431915730 -1.395523790 CARBON 6.0 -1.166676574 0.550508040 -0.705577944 CARBON 6.0 -1.166676574 0.550508040 0.705577944 CARBON 6.0 -0.486294808 -0.431915730 1.395523790 HYDROGEN 1.0 -0.352988862 -0.347708199 2.465648783
IDROGENO 1,0 -0,084717264 -1,289523730 0,864198908 IDROGENO 1,0 -1,712490163 1,338624758 1,223514576 IDROGENO 1,0 -1,712490163 1,338624758 -1,223514576 IDROGENO 1,0 -0,352988862 -0,347708199 -2,465648783 IDROGENO 1,0 -0,084717264 -1,289523730 -0,864198908 CARBONIO 6,0 1,559305232 -0,085874351 -0,690651129 CARBONIO 6,0 1,559305232 -0,085874351 0,690651129 IDROGENO 1.0 1.514911897 -1.022640981 1.232100569 HYDROGEN 1.0 1.603704743 0.851022589 1.232175888 HYDROGEN 1.0 1.603704743 0.851022589 -1.232175888 HYDROGEN 1.0 1.514911897 -1.022640981 -1.232100569 $ END
$ END
e nel mio esempio sono stati eseguiti 19 passaggi.

Ora è possibile utilizzare i risultati di questo calcolo per eseguire una ricerca completa dello stato di transizione. Ricorda che hai tutte le informazioni necessarie per riavviare una corsa nell'output ausiliario, cioè il file .dat . Dovresti sempre conservare questo fino a quando non hai completato i calcoli.
Se vuoi fare calcoli e chiederti quale sia la geometria ottimizzata, ecco i risultati del passaggio finale:
----- RISULTATI DELLA RICERCA GEOMETRICA DI RHF RIUSCITA ---------- COORD, ORB, GRADIENTE E APPROSS. HESSIAN ----- COORDINATE DI SIMMETRIA ATOMI UNICI (ANGS) CARICA ATOMICA XYZ --------------------------------- --------------------------- CARBON 6.0 -0.4847330011 -0.4122786646 -1.4086312155 CARBON 6.0 -1.1902020846 0.5288839653 -0.7019679480 CARBON 6.0 -1.1902020983 0.5288839622 0.7019679173 CARBON 6.0 -0.4847329728 -0.4122786820 1.4086312270 IDROGENO 1.0 -0.4076405265 -0.3274824211 2.4860530248
IDROGENO 1,0 -0,4019303863 -1,4244097276 1,0396872383 IDROGENO 1,0 -1,5467259917 1,4139143889 1,2140730563 IDROGENO 1,0 -1,5467259527 1,4139143906 -1,2140730901 IDROGENO 1,0 -0,4076405381 -0,3274824126 -2,4860529903 IDROGENO 1,0 -0,4019304494 -1,4244096950 -1,0396872193 CARBONIO 6,0 1,5587691569 -0,0858507824 -0,6884831136 CARBONIO 6,0 1,5587691834 -0,0858507803 0,6884831288 IDROGENO 1.0 1.9167366148 -0.9561957837 1.2206602417 IDROGENO 1.0 1.6758814772 0.8473264236 1.2212666939 IDROGENO 1.0 1.6758814263 0.8473264124 -1.2212666800 IDROGENO 1.0 1.9167365785 -0.942 Thu52: 7775 RH -1.220660 GENERATO -0.9561957779 DICEMBRE -067 (NUC) = 227.2020852580 $ VEC ...
Dichiarazione di non responsabilità : esiste un altro modo per bloccare le coordinate. Potrei aggiungerlo in un altro punto come un'altra risposta. È un po 'più complicato, tuttavia offre anche una maggiore flessibilità.