La domanda chiede perché l'acqua ha un angolo maggiore di altri idruri della forma $ \ ce {XH2} $ in particolare $ \ ce {H2S} $ e $ \ ce {H2Se} $. Ci sono state altre domande simili, quindi di seguito viene fornito un tentativo di una risposta generale.
Ci sono, ovviamente, molti altri idruri triatomici, $ \ ce {LiH2} $, $ \ ce {BeH2} $, $ \ ce {BeH2} $, $ \ ce {NH2} $, ecc. Risulta che alcuni sono lineari e altri a forma di V, ma con angoli di legame differenti, e che la stessa spiegazione generale può essere usata per ciascuno di questi casi .
È chiaro che, poiché l'angolo di legame per l'acqua non è né $ 109,4 ^ \ circ $, $ 120 ^ \ circ $, né $ 180 ^ \ circ $ che $ \ ce {sp ^ 3} $, $ L'ibridazione \ ce {sp ^ 2} $ o $ \ ce {sp} $ non spiegherà gli angoli di legame. Inoltre, lo spettro del fotoelettrone UV dell'acqua, che misura le energie orbitali, deve essere spiegato così come gli spettri di assorbimento UV.
La via d'uscita da questo problema è fare appello alla teoria degli orbitali molecolari e costruire orbitali basati sugli orbitali $ \ ce {s} $ e $ \ ce {p} $ e sulla loro sovrapposizione al variare dell'angolo di legame. Il diagramma orbitale è stato elaborato molto tempo fa è ora chiamato diagramma di Walsh (AD Walsh J. Chem. Soc. 1953, 2262; DOI: 10.1039 / JR9530002260). La figura seguente mostra un diagramma di questo tipo e i prossimi paragrafi spiegano la figura.
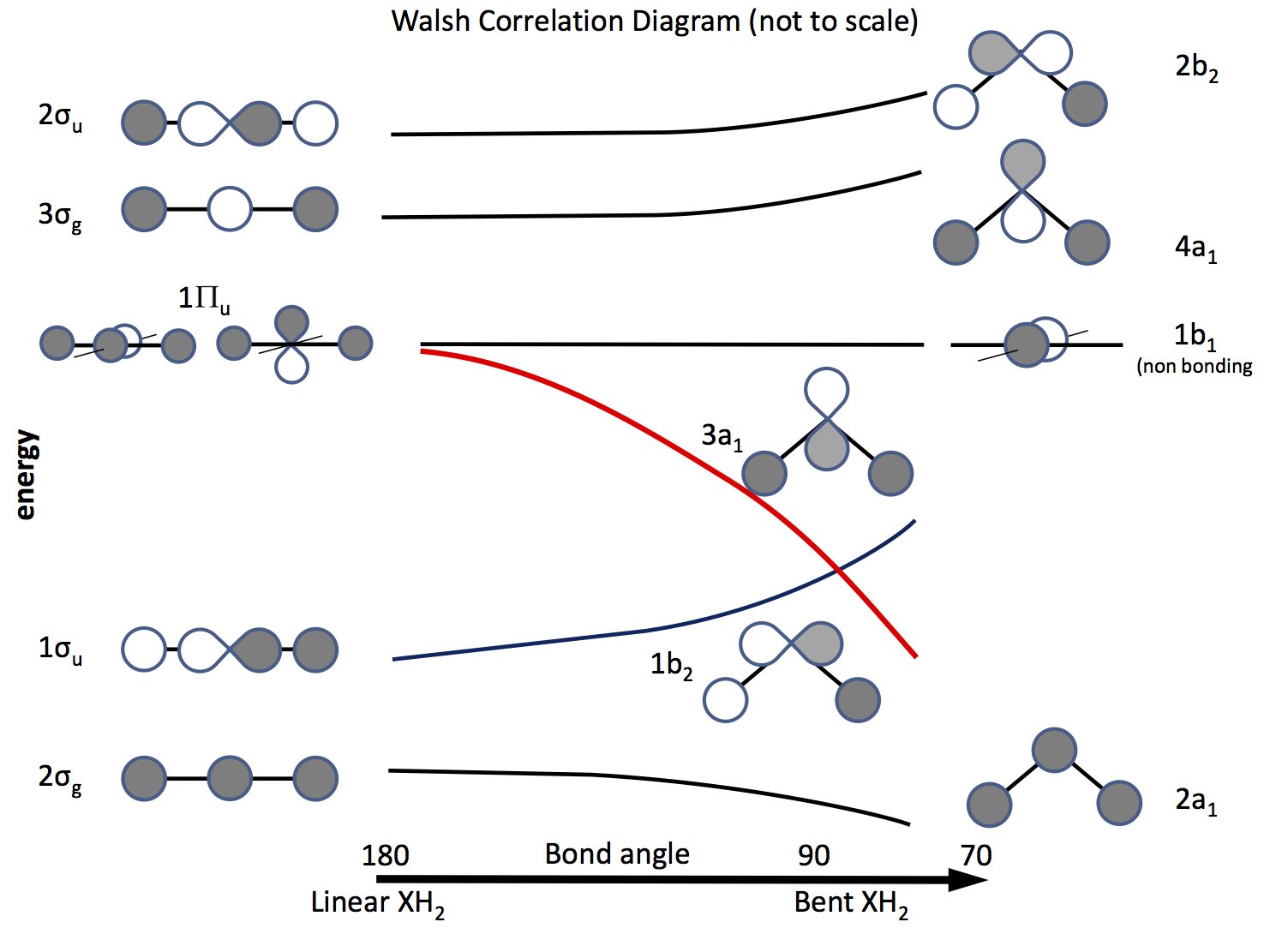
L'ombreggiatura indica il segno (fase) dell'orbitale, "mi piace" si sta legando altrimenti non legandosi. Le energie sono relative così come la forma delle curve. A sinistra ci sono gli orbitali disposti in ordine di energia crescente per una molecola lineare; a destra quelli per una molecola piegata. Gli orbitali etichettati $ \ Pi_ \ mathrm {u} $ sono degeneri nella molecola lineare ma non così in quelli piegati. Le etichette $ \ sigma_ \ mathrm {u} $, $ \ sigma_ \ mathrm {g} $ si riferiscono ai legami sigma, gli indici $ \ mathrm {g} $ e $ \ mathrm {u} $ si riferiscono al fatto che il MO combinato abbia un centro di inversione $ \ mathrm {g} $ (gerade) o no $ \ mathrm {u} $ (ungerade) e derivano dalle rappresentazioni irriducibili nel gruppo $ D_ \ mathrm {\ infty h} $ punti. Le etichette sul lato destro si riferiscono alle rappresentazioni nel gruppo $ C_ \ mathrm {2v} $ punti.
Dei tre $ \ Pi_ \ mathrm {u} $ orbitali uno forma il $ \ sigma_ \ mathrm {u} $, gli altri due sono degeneri e non leganti .
Uno degli $ \ ce {p} $ orbitali si trova nel piano del diagramma, l'altro fuori l'aereo, verso il lettore.
Quando la molecola viene piegata, questo orbitale rimane senza legami, l'altro diventa $ \ ce {3a_1} $ orbitale (linea rossa) la cui energia si abbassa notevolmente quando si sovrappone con gli aumenti orbitali dell'atomo H.
Per capire se una molecola è lineare o piegata, tutto ciò che serve è mettere elettroni negli orbitali. Quindi, la prossima cosa è fare un elenco del numero di elettroni possibili e vedere cosa prevede il diagramma. \ Begin {array} {rcll} \ text {Nr.} & \ text {Shape} & \ text {molecule (s) } & \ text {(angolo, configurazione)} \\ \ hline 2 & \ text {bent} & \ ce {LiH2 +} & (72, ~ \ text {calcolato}) \\ 3 & \ text {linear} & \ ce {LiH2}, \ ce {BeH2 +} & \\ 4 & \ text {linear} & \ ce {BeH2}, \ ce {BH2 +} & \\
5 & \ text {bent} & \ ce {BH2} & (131, \ ce {[2a_1 ^ 2 1b_2 ^ 2 3a_1 ^ 1]}) \\ 6 & \ text {bent} & \ ce {^ 1CH2} & (110, \ ce {[1b_2 ^ 2 3a_1 ^ 2]}) \\ & & \ ce {^ 3CH2} & (136, \ ce {[1b_2 ^ 2 3a_1 1b_1 ^ 1]}) \\ & & \ ce {BH2 ^ -} & (102) \\ & & \ ce {NH2 +} & (115, \ ce {[3a_1 ^ 2])} \\ 7 & \ text {bent} & \ ce {NH2} & (103,4 , \ ce {[3a_1 ^ 2 1b_1 ^ 1]}) \\ 8 & \ text {bent} & \ ce {OH2} & (104.31, \ ce {[3a_2 ^ 2 1b_1 ^ 2]}) \\ & & \ ce {NH2 ^ -} & (104) \\ & & \ ce {FH2 ^ +} & \\ \ hline \ end {array}
Altri idruri mostrano effetti simili a seconda del numero di elettroni in $ \ ce {b2} $, $ \ ce {a1} $ e $ \ ce {b1} $ orbitali; ad esempio: \ begin {array} {ll} \ ce {AlH2} & (119, \ ce {[b_2 ^ 2 a1 ^ 1]}) \\ \ ce {PH2} & (91.5, \ ce {[b_2 ^ 2 a_1 ^ 2 b_1 ^ 1]}) \\ \ ce {SH2} & (92) \\ \ ce {SeH2} & (91) \\ \ ce {TeH2} & (90.2) \\ \ ce {SiH2} & (93) \\\ end {array}
L'accordo con l'esperimento è qualitativamente buono, ma, ovviamente, gli angoli di legame non possono essere determinati con precisione con un modello di base solo tendenze generali.
Lo spettro fotoelettronico (PES) dell'acqua mostra segnali da $ \ ce {2a1} $, $ \ ce {1b2} $, $ \ ce {3a1} $, $ \ ce {1b1} $ orbitali, ($ 21,2 $ , $ 18,7 $, $ 14,23 $ e $ \ pu {12,6 eV} $ rispettivamente) l'ultimo è non vincolante, come mostrato dalla mancanza di struttura. I segnali dagli orbitali $ \ ce {3b2} $ e $ \ ce {3a1} $ mostrano una struttura vibrazionale che indica che questi sono orbitali di legame.
L'intervallo di assorbimento UV e visibile di $ \ ce {BH2} $, $ \ ce {NH2} $, $ \ ce {OH2} $ è $ 600 - 900 $, $ 450 - 740 $ e $ 150 - \ pu {200 nm} $ rispettivamente. $ \ ce {BH2} $ ha un piccolo divario energetico HOMO-LUMO tra $ \ ce {3a1} $ e $ \ ce {1b1} $ poiché lo stato fondamentale è leggermente piegato. Si prevede che il primo stato eccitato sia lineare poiché la sua configurazione è $ \ ce {1b_2 ^ 2 1b_1 ^ 1} $ e questo viene osservato sperimentalmente.
$ \ ce {NH2} $ ha un HOMO- Divario energetico LUMO da $ \ ce {3a_1 ^ 2 1b_1 ^ 1} $ a $ \ ce {3a_1 ^ 1 1b_1 ^ 2} $, quindi sia lo stato fondamentale che quello eccitato dovrebbero essere piegati, l'angolo dello stato eccitato è di circa $ 144 ^ \ circ $. Rispetto a $ \ ce {BH2} $, $ \ ce {NH2} $ è più piegato quindi il divario energetico HOMO-LUMO dovrebbe essere maggiore come osservato.
$ \ ce {OH2} $ ha un HOMO -LUMO energy gap da $ \ ce {3a_1 ^ 2 1b_1 ^ 2} $ a $ \ ce {3a_1 ^ 2 1b_1 ^ 1 4a_1 ^ 1} $, cioè un elettrone promosso dall'orbitale di non legame al primo anti-legame orbitale. La molecola eccitata rimane piegata in gran parte a causa del forte effetto di due elettroni in $ \ ce {3a1} $ che contrastano il singolo elettrone in $ \ ce {4a1} $. L'angolo di obbligazione è quasi invariato a $ 107 ^ \ circ $, ma il divario energetico sarà maggiore che in $ \ ce {BH2} $ o $ \ ce {NH2} $, sempre come osservato.
Gli angoli di legame di $ \ ce {NH2} $, $ \ ce {NH2 -} $ e $ \ ce {NH2 +} $ sono tutti molto simili, $ 103 ^ \ circ $, $ 104 ^ \ circ $ e $ 115 ^ \ circ $ rispettivamente. $ \ ce {NH2} $ ha la configurazione $ \ ce {3a_1 ^ 2 1b_1 ^ 1} $ dove $ \ ce {b1} $ è un orbitale non legante, quindi l'aggiunta di un elettrone fa poca differenza, rimuoverne uno significa che il $ \ ce {3a_1} $ orbitale non è stabilizzato tanto e quindi l'angolo di legame è leggermente aperto.
Le molecole di stato $ \ ce {CH2} $ di singoletto e tripletto mostrano che il singoletto ha due elettroni nell'orbitale $ \ ce {3a1} $ e ha un angolo più piccolo dello stato di tripletto con un solo elettrone qui e uno nel $ \ ce {b1} $ non legame, quindi l'angolo di legame allo stato fondamentale della tripletta dovrebbe essere più grande del singoletto.
Man mano che la dimensione dell'atomo centrale aumenta, il suo nucleo diventa più schermato dagli elettroni del nucleo e diventa meno elettronegativo. Così scendendo nella tavola periodica il legame $ \ ce {XH} $ diventa meno ionico, più densità di elettroni è intorno all'atomo $ \ ce {H} $ quindi il nucleo $ \ ce {H} $ è meglio schermato, e quindi il $ \ ce {XH} $ bond è più lungo e più debole. Pertanto, come al solito con le tendenze all'interno della stessa famiglia nella tavola periodica, l'effetto è, fondamentalmente, di dimensioni atomiche.
Le molecole con un atomo centrale più pesante, $ \ ce {SH2} $, $ \ ce {PH2} $, ecc. hanno tutte angoli di legame intorno a $ 90 ^ \ circ $. La diminuzione dell'elettronegatività destabilizza l'orbitale $ \ Pi_ \ mathrm {u} $ aumentandone l'energia. Gli orbitali $ \ ce {s} $ degli atomi centrali più pesanti sono più grandi e di minore energia di quelli dell'ossigeno, quindi questi orbitali si sovrappongono con l'orbitale $ \ ce {s} $ dell'atomo $ \ ce {H} $ più debolmente. Entrambi questi fattori aiutano a stabilizzare l'orbitale $ 3 \ sigma_ \ mathrm {g} $ lineare e quindi il $ \ ce {4a1} $ nella configurazione piegata. Questo orbitale appartiene alla stessa specie di simmetria di $ \ ce {3a1} $ e quindi possono interagire mediante un'interazione Jahn-Teller del secondo ordine. Questo è proporzionale a $ 1 / \ Delta E $ dove $ \ Delta E $ è il divario di energia tra i due orbitali menzionati. L'effetto di questa interazione è di aumentare $ \ ce {4a1} $ e diminuire $ \ ce {3a1} $ in energia. Quindi, scendendo nella serie $ \ ce {OH2} $, $ \ ce {SH2} $, $ \ ce {SeH2} $, ecc., L'angolo di legame dovrebbe diminuire, come si osserva.
Sono stati forniti esempi per $ \ ce {XH2} $ molecole, ma questo metodo è stato utilizzato anche per comprendere le molecole triatomiche e tetra-atomiche in generale, come $ \ ce {NO2} $, $ \ ce {SO2} $, $ \ ce {NH3} $, ecc.