Presenterò un argomento LCAO-MO. Ma prima sfatiamo un mito.
$ \ ce {SF6} $ ha zolfo "ipervalente" e gli orbitali 3d sullo zolfo partecipano al legame
No. Questo non è vero. Chiuderei un occhio se viene insegnato al liceo, ma davvero, questo non può essere più lontano dalla verità. Ecco un riferimento: J. Am. Chem. Soc. 1986, 108, 3586; ce ne sono molti altri. Per coloro che vogliono saperne di più, c'è una grande quantità di informazioni sulla controversia sulla regola dell'orbitale-d e dell'ottetto; una semplice ricerca sul sito web di ACS per "ipervalenza" produrrà una serie di risultati. Purtroppo non sono riuscito a trovare una recensione sull'argomento.
Comunque, come si descrive la struttura di $ \ ce {SF6} $? Ecco un "semplice" diagramma MO (non passerò attraverso i dettagli su come costruirlo). In realtà è abbastanza simile a quello di un complesso metallico di transizione ottaedrico, tranne per il fatto che qui gli orbitali 3s e 3p sullo zolfo sono al di sotto degli orbitali 3d.

Solo per il gusto di contare gli elettroni, ho trattato il composto come "completamente ionico", cioè $ \ ce {S ^ 6 +} + 6 \ ce {F -} $. Quindi lo zolfo ha iniziato con 0 elettroni di valenza e ogni fluoro ha iniziato con 2 elettroni nei suoi orbitali σ. Ho anche trascurato il contributo del π al legame, quindi le coppie solitarie del fluoro non appaiono nel diagramma.
Vedrai che, per un totale di sei $ \ ce {SF} $ legami , abbiamo solo quattro coppie di elettroni per legare gli MO. Le altre due coppie di elettroni sono localizzate sui $ \ mathrm {e_g} $ MO, che non si legano e sono localizzati sul fluoro. Se vogliamo assegnare una carica formale allo zolfo in base a questo diagramma, sarebbe +2, perché in realtà ci sono solo quattro legami. Potremmo forse usare i diagrammi di Lewis per rappresentarlo in questo modo:
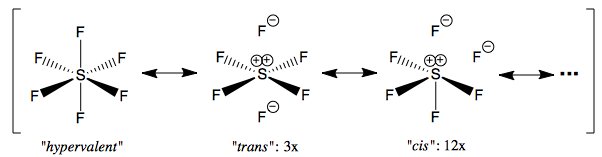
La forma di risonanza "ipervalente" contribuisce piuttosto poco e non si basa sull'invocazione della partecipazione d-orbitale; vedere il commento di Martin sulla mia risposta di seguito per maggiori dettagli sui contributi di risonanza. Immagino che la sua esistenza possa essere principalmente attribuita a iperconiugazione negativa, anche se non ne sono sicuro al 100%. Le forme di risonanza trans e cis non sono uguali, quindi il loro contributo non è lo stesso, ma il contributo di ciascuna forma di risonanza trans individuale deve essere lo stesso per simmetria. Complessivamente, i sei fluoro in $ \ ce {SF6} $ devono essere equivalenti per la simmetria ottaedrica della molecola. Potresti eseguire un $ \ ce {^ 19F} $ NMR del composto e dovrebbe darti solo un picco.
(Un modo alternativo di vederlo è che due dei $ \ ce {SF } Le obbligazioni $ sono "vere" obbligazioni 2c2e e che le altre quattro obbligazioni $ \ ce {SF} $ "sono in realtà solo un paio di obbligazioni 3c4e, ma non ne parlerò. Per maggiori informazioni su multi- center bond, questo articolo è una bella introduzione: J. Chem. Educ. 1998, 75, 910; vedere anche i riferimenti 12 e 13 in quell'articolo.)
Fin dall'inizio, possiamo vedere perché $ \ ce {SH6} $ non è tanto favorito. Se usiamo la stessa struttura per descrivere il legame in $ \ ce {SH6} $, allora quelle forme di risonanza "corrette" che abbiamo disegnato coinvolgerebbero $ \ ce {H -} $. Lascio al lettore capire se $ \ ce {F -} $ o $ \ ce {H -} $ è più stabile.
In alternativa, se vuoi restare fedele al MO descrizione, l'idea è che in $ \ ce {SH6} $, l'energia relativamente alta di H1s rispetto a F2p porterà gli orbitali $ \ mathrm {e_g} $ non legati ad essere relativamente più alti in energia. A parità di condizioni, è meno favorevole che venga occupato un orbitale di maggiore energia, e $ \ ce {SH6} $ sarebbe quindi molto incline a perdere questi elettroni, cioè a essere ossidato.
In effetti, se rimuoviamo quei quattro elettroni dagli orbitali $ e_ \ mathrm {g} $, è possibile che si formino questi idruri a sei coordinate. Ma ovviamente potremmo non voler avere una molecola $ \ ce {SH6 ^ 4 +} $ a piede libero. Probabilmente perderà tutti i suoi protoni in fretta e furia per tornare a essere $ \ ce {H2S + 4H +} $. C'è qualcosa di meglio? Ebbene, c'è la specie $ \ ce {CH6 ^ 2 +} $, che è protonata due volte dal metano. È isoelettronico di valenza con $ \ ce {SH6 ^ 4 +} $, e se vuoi leggerlo, ecco un articolo: J. Am. Chem. Soc. 1983, 105, 5258. Anche se non è certo la molecola più stabile del pianeta, è sicuramente già molto meglio di $ \ ce {SH6} $.
Ora, solo per tornare al punto da cui siamo partiti: la partecipazione d-orbitale. Sì, esiste un $ \ mathrm {e_g} $ insieme di orbitali d che può sovrapporsi alla combinazione $ \ mathrm {e_g} $ lineare di orbitali F2p apparentemente "non vincolanti", stabilizzandola in tal modo. "Ah! Ci hai mentito per tutto questo tempo! Gli orbitali d sono importanti!" No. Aiutano, ma quanto aiutano effettivamente ? Niente affatto, considerando il grande divario di energia tra gli orbitali S3d e F2p - di nuovo, vedi i commenti di Martin per maggiori dettagli.
Come ho mostrato, non hai bisogno di orbitali d per razionalizzare 6 -coordina lo zolfo, e non hai bisogno di d orbitali per spiegare il fosforo a 5 coordinate o la forza del legame $ \ ce {P = O} $. L'unico motivo per cui si dovrebbero mai disegnare strutture di Lewis in quanto tali è per semplicità.