Sì, la molecola dovrebbe mostrare chiralità al di sotto della temperatura ambiente.
Nella configurazione molecolare ottimizzata 1 la molecola non può essere sovrapposta alla sua immagine speculare. Ciò è dovuto alla rotazione fuori dal piano dei gruppi nitro.
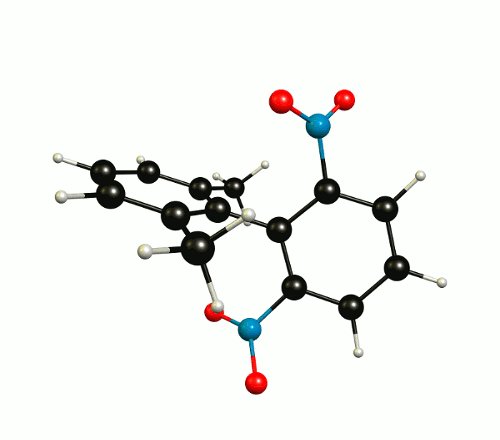
La struttura termodinamica più favorevole è di $ C_2 $ simmetria e non ha un piano speculare (vedi sopra).
Contrariamente agli argomenti presentati in precedenza, un'analisi della struttura di Lewis è fuorviante in quanto è in modo massicciamente errata il requisito di spazio delle frazioni nitro. Una caratteristica che è già presente nel 2,4,6-trinitrotoluene. 2 Dall'abstract di Clarkson et.al. : 3
Sono state trovate due autentiche strutture di minimo energetico. In entrambe le strutture il gruppo 4-nitro è planare all'anello fenile, mentre i gruppi 2,6-nitro sono leggermente fuori piano con l'anello fenilico a causa dell'interazione sterica con il gruppo metile.
Successivamente descrivono:
Le due strutture molecolari stabili di TNT, Fig. 1, sono correlate da rotazioni interne dei gruppi 2 e 6-nitro e del gruppo metile. [... ] La struttura A è la geometria più stabile, anche se la differenza di energia è piccola ( $ 0.650 ~ \ pu {kcal mol ^ −1} $ in B3LYP / 6-311 + G **, Tabella 4). La struttura A mostra una simmetria quasi ideale $ C_ \ mathrm {s} $ con uno degli atomi di idrogeno metilico perpendicolare all'anello fenilico nel piano di $ \ sigma_ \ mathrm {h} $ . I gruppi 2,6-nitro di A sono non planari e ruotati nella stessa faccia dell'anello fenilico, massimizzando il numero di interazioni di van der Waals con il gruppo metile.
La frazione 2,6-fenile tuttavia è molto più grande di un gruppo metile, motivo per cui la simmetria dei gruppi nitro è rotta. Forzare questa $ C_ \ mathrm {s} $ simmetria porterà a un punto di sella del primo ordine (stato di transizione) sulla superficie energetica potenziale che è solo di circa $ 1,6 ~ \ pu {kJ mol ^ -1} $ maggiore di energia rispetto alla struttura minima.
A temperatura ambiente è probabile che l'interconversione tra gli enantiomeri avvenga attraverso questo struttura. Non ho verificato se questo stato si connette effettivamente tramite un calcolo di coordinate di reazione intrinseca, ma dall'indagine visiva della modalità immaginaria questo sembra molto probabile.

Se raffreddi abbastanza il sistema dovresti essere in grado di identificare due enantiomeri, tuttavia, a temperatura ambiente, l'interconversione dovrebbe essere troppo veloce per essere osservata.

Sarebbe utile sapere quale libro hai utilizzato e in quale contesto è stata rilasciata la dichiarazione. Da un punto di vista puramente teorico, sarei d'accordo sul fatto che la molecola è chirale. Dal punto di vista pratico non valuterei questa proprietà, poiché probabilmente è troppo difficile da misurare.
Il $ C_ \ mathrm {2v suggerito Le strutture} $ sono punti di sella del secondo o del quarto ordine, a seconda della rotazione dei gruppi metilici. Queste strutture sono almeno $ 15 ~ \ pu {kJ mol ^ -1} $ più elevate di energia rispetto alle prime e non sono un percorso percorribile per l'interconversione.

Footnotes
- Tutte le strutture sono state ottimizzate a livello di teoria DF-M06L / def2-SVP con gaussiano 09 Rev. D01. I punti stazionari sono stati caratterizzati calcolando le frequenze vibrazionali allo stesso livello di teoria.
- W. Robert Carper, Larry P. Davis, Michael W. Extine, J. Phys. Chem. 1982, 86 (4), 459–462.
- John Clarkson, W. Ewen Smith, David N. Batchelder, D. Alastair Smith, Alison M. Coats, J. Mol. Struc. 2003, 648 (3), 203-214.